Was ist ein GIST?
WAS IST EIN GIST? GIST (Gastrointestinale Stromatumore) sind selten auftretende Weichgewebstumore des Gastrointestinaltrakts (Magen-Darmtrakt). Die Gruppe der Weichgewebstumore umfasst gutartige und bösartige (sog. Sarkome) Tumore, die sich im Binde- und Stützgewebe wie z. B. Muskelgewebe oder Knochen entwickeln. Im Gegensatz zu den häufig auftretenden bösartigen, epithelialen Tumoren, den sog. Karzinomen, sind Sarkome äußerst selten und machen lediglich 1 % der bösartigen Tumorerkrankungen aus. Bedingt durch die Seltenheit dieser Erkrankung gibt es nur wenige Onkologen, Pathologen, Chirurgen, Radiologen etc., die sich auf die Behandlung von PatientInnen mit einem Sarkom spezialisiert haben.
WIE HÄUFIG IST GIST? Pro Jahr erkranken etwa 15 Personen pro 1 Million EinwohnerInnen an einem klinisch relevanten GIST (in Österreich etwa 120 Neuerkrankungen pro Jahr). Etwa die Hälfte weisen bei Diagnosestellung Metastasen (Tochtergeschwülste) bevorzugt in der Leber oder im Bauchraum auf. Klinisch stumme (asymptomatische) Mikro-GIST (2–10 mm) sind jedoch deutlich häufiger und konnten bei bis zu 30 % von PatientInnen über 70 Jahre nachgewiesen werden.
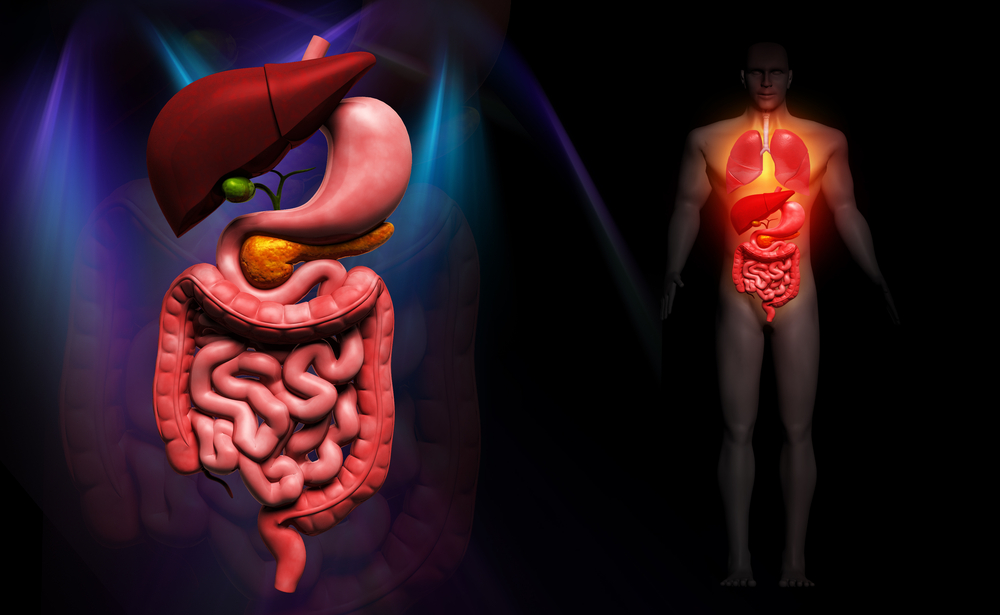
WO KOMMT GIST VOR? GIST treten im gesamten Gastrointestinaltrakt auf, am häufigsten im Magen (gastro), ca. 50–70 %, gefolgt vom Dünndarm (intestinal), ca. 20–30 %. Selten treten GIST auch in der Speiseröhre, im Zwölffingerdarm, Dickdarm und Mastdarm auf.
WIE LANGE KENNT MAN SCHON GIST? Als gut definierte, gut diagnostizierbare und in Folge therapierbare Tumorentität existiert der GIST seit 1998.
WARUM ENTSTEHT EIN GIST? Ursache für die Entstehung eines GIST sind Genmutationen. Das menschliche genetische Material (Erbmaterial/Erbinformation) besteht aus ca. 30.000 verschiedenen Genen. Jedes dieser Gene ist in der DNA (Substanz, die die genetische Information beherbergt) in jeder Zelle des Körpers vorhanden. Gene sorgen dafür, dass die Zelle Baupläne für die Produktion von Proteinen (Eiweiße) zur Verfügung hat, die für das Überleben von Zellen, den Aufbau von Zellen und die Kommunikation zwischen den Zellen notwendig sind.
WELCHE MUTATIONEN SIND DAS? Bei GIST sind hauptsächlich Mutationen im KIT-Gen (ca. 80–85 %) und PDGFRA-Gen (5–10 %) bekannt. Unter 10 % der GIST weisen keine Mutationen in KIT und PDGFRA auf und werden als „wild-typ“ bezeichnet. Selten wurden in diesen wild-typ GIST Mutationen im BRAF-Gen (7–9 %) und SDH B-, C- oder D-Gen nachgewiesen. Bedingt durch die Mutation werden die Rezeptortyrosinkinase KIT oder PDGFRA falsch zusammengesetzt und dadurch bedingt in der Zelle Prozesse aktiviert, die eine ungehinderte Zellteilung bzw. ein ungehindertes Zellwachstum bewirken.
KIT- und PDGFRA-Mutationen treten nur in bestimmten Abschnitten (Exons) des KIT- und PDGFRA-Gens auf („hot spots“). Im KIT-Gen finden sich die Mutationen in Exon 9 (9 %), Exon 11 (70 %), Exon 13 (1 %) und Exon 17 (1 %). Im PDGFRA-Gen finden sich die Mutationen in Exon 12 (<1 %), Exon 14 (<1 %) und Exon 18 (8 %). Die Lokalisation der Genmutation gibt Aufschluss über das Therapieansprechen (siehe Therapie). Bedingt durch die Entdeckung dieser Mutationen konnten auch Aufschlüsse über die Ursprungszelle der GIST, Biomarker für die GIST-Diagnostik (siehe Absatz GIST-Diagnostik) und Ansatzpunkte für eine zielgerichtete („targeted“) Therapie gewonnen werden.
WO IST DIE URSPRUNGSZELLE DES GIST? Bedingt durch die Erkenntnisse der modernen Medizin und den Nachweis von Biomarkern an Tumorgewebe konnte man zeigen, dass GIST sich nicht, wie historisch angenommen, von der glatten Muskulatur des Gastrointestinaltraktes (GI-Trakt) ableiten, sondern Ähnlichkeiten mit den Schrittmacher-Zellen (Pacemaker-Cells) des GI-Trakts, den interstitiellen Zellen von Cajal, haben. Diese Zellen sind zwischen der glatten Muskulatur des GI-Trakts lokalisiert und für die Peristaltik (Fortbewegung von Nahrung durch den GI-Trakt) verantwortlich. Der GIST entsteht daraus resultierend in der Wand des GI-Traktes.

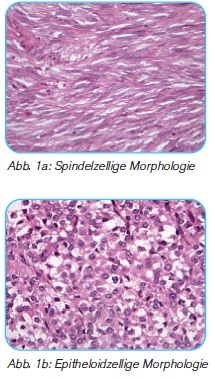
WAS MACHT DER PATHOLOGE? Die feingewebliche/mikroskopische Untersuchung von Tumorgewebe ist sowohl unerlässlich für die Bestimmung der Tumorentität als auch für die Risikoeinschätzung. Die Informationen, die der Pathologe liefert, sind essentiell für die Therapieplanung und Behandlung einer/eines Patientin/Patienten.
Nach Einlangen einer Gewebsprobe (Biopsie) oder eines Tumoroperationspräparats (gesamter Tumor) wird das Tumorgewebe (Tumorzellen) unter dem Mikroskop betrachtet (siehe Bilder). Die Tumorlokalisation, Form der Tumorzellen und das Wachstumsmuster helfen dem Pathologen einen GIST zu erkennen. Um diesen Tumor auch eindeutig als GIST zu bestätigen, werden Spezialfärbungen (Immunhistochemie genannt) durchgeführt. Mit Antikörpern gegen KIT und DOG1(für GIST typische Biomarker) zeigt das Tumorgewebe eine deutliche Anfärbbarkeit (Reaktivität). Zusätzlich kann der Pathologe am vorliegenden Tumorgewebe auch KIT- oder PDGFRA-Mutationen bestimmen. Diese Mutationsanalysen sind dann von Bedeutung, wenn eine medikamentöse Therapie geplant ist (z. B. chirurgisch nicht entfernbare und metastasierte GIST, primäre GIST mit hohem Risiko auf ein Rezidiv (Rückfall).
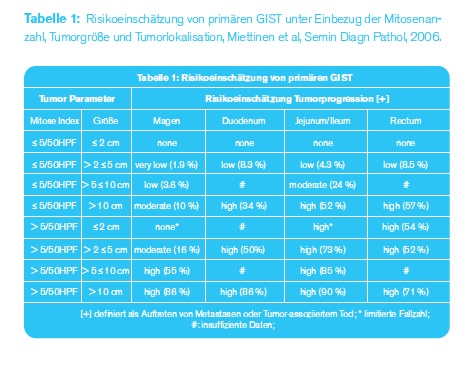
IST JEDER GIST BÖSARTIG? Die Risikoeinschätzung eines primären GIST obliegt dem Pathologen und es werden heute die sogenannten Miettinenkriterien herangezogen. Unter Einbezug der Tumorlokalisation, Tumorgröße und Anzahl der Mitosen (Kernteilungsfiguren) pro 50/HPF (10 mm2) erfolgt die Risikoeinschätzung durch den Pathologen. Gundsätzlich kann man sagen, dass unabhängig von der Lokalisation Tumore kleiner oder gleich 2 cm als gutartig eingestuft werden können.
GIBT ES NOCH ANDERE ARTEN VON GIST?
- PatientInnen mit Neurofibromatose Typ 1 (NF-1) können in 25 bis 30 % der Fälle mehrere, überwiegend kleine GIST bevorzugt im Dünndarm aufweisen. Die kleinen GIST sind in der Regel gutartig. Die Tumore zeigen eine immunhistochemische Reaktivität mit KIT sowie DOG-1 auf und weisen in der Regel keine KIT- und PDGFRA-Mutationen auf.
- Pädiatrische GIST (PGIST oder GIST im Kindesalter) treten bevorzugt in der zweiten Lebensdekade auf und machen etwa 1 bis 2 % aller GIST aus. PGIST treten bevorzugt bei Frauen auf und sind überwiegend im Magen lokalisiert. Oft treten auch mehrere GIST im Magen auf. Im Unterschied zu GIST des Erwachsenen muss hervorgehoben werden, dass PGIST auch in Lymphknoten metastasieren. Pädiatrische GIST zeigen eine immunhistochemische Reaktivität mit KIT sowie DOG-1 und weisen in der Regel keine KIT- und PDGFRA-Mutationen auf. PGIST unterscheiden sich deutlich von den GIST des Erwachsenen und werden heute als eigenständige Tumorentität angesehen. Speziell in dieser Patientengruppe sollten mit PGIST assozierte Syndrome (Carney´s Trias, Carney-Stratakis-Syndrom und seltener NF1 und familiäre GIST) ausgeschlossen werden. PGIST weisen gehäuft SDHB- oder C-Mutationen auf.
IST GIST VERERBBAR? Familiäre GIST-Syndrome treten in Folge einer Keimbahnmutation im KIT- oder PDGFRA-Gen auf und werden vererbt. Familiäre GIST sind äußerst selten mit derzeit etwa 22 dokumentierten Familien weltweit. Die meisten betroffenen Familienmitglieder entwickeln einen oder mehrere GIST während ihres Lebens. Der deutliche Unterschied von familiären GIST zu sporadischen GIST ist dadurch gekennzeichnet, dass familiäre GIST an mehreren Stellen des Gastrointestinaltrakts auftreten und mit anderen Merkmalen wie z. B. Auftreten von Pigmentflecken an den Händen und perianal, Muttermalen, gutartigen Weichteiltumore einhergehen.
– Dr. Thomas Brodowicz, Medizinische Universität Wien